
二谱法品添超高测定中的种食效液相色同时加剂饮料
(4)流速的超高测定选择
流速的改变会影响梯度洗脱时间和分离效果。试验考察了0.10~0.60mL/min范围间10种目标化合物的效液相色分离效果。当流速大于0.2mL/min时,谱法品添出峰时间提前,饮料容易造成各目标物之间分离度和峰型都不理想。中的种食当流速小于0.15mL/min时,加剂延长了目标物出峰时间,超高测定降低了工作效率。效液相色所以,谱法品添本试验选用0.15mL/min作为方法最佳流速。饮料
(5)梯度洗脱程序的中的种食选择
试验分析了流动相中甲醇初始比例对分离度的影响。当甲醇比例大20%时,加剂前4种目标化合物出峰较快,超高测定但色谱峰响应值低,效液相色易出现色谱峰重叠现象;当甲醇比例小于20%时,谱法品添各目标物出峰时间延长,并且峰形差,所以选择甲醇的初始比例为20%,0.02mmol/L乙酸铵的初始比例为80%。梯度洗脱程序运行到10min时,甲醇比例再重新回到20%,目的是稳定基线,方便下一个目标物实现更好的分离。

(6)柱温的选择
本试验考察了柱温在20℃~50℃范围间的检测结果,当T<30℃时,各组分分离效果较差1当T>30℃时,柱效降低。因此,选择30℃为最佳检测柱温温度。
在优化的实验条件下,10种食品添加剂在10min内能满足快速分离的需要,且分离度与灵敏度都较好。标准色谱图见图4。

三、标准曲线、检出限及精密度测定
1、标准曲线、检出限的测定
按优化好的试验条件对10种食品添加剂混合标准溶液系列进行色谱分析,以各组分的峰面积为纵坐标,质量浓度为横坐标,并绘制标准曲线。结果表明,在0.050~50.0μg/mL范围内,10种食品添加剂的质量浓度与峰面积呈良好的线性关系,相关系数R2均大于0.999,以3倍信噪比(3S/N)计算方法检出限,方法检出限介于0.0017~0.0034μg/mL之间。标准曲线、相关系数、检出限及线性范围见表2。

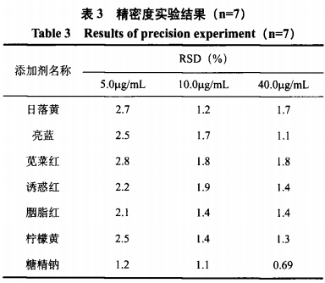
2、精密度试验
本试验分别对5.0μg/mL、10.0μg/mL和40.0μg/mL三组浓度的混合标准溶液进行7次平行测定。如表3所示,测定低、中、高三组浓度的相对标准偏差(RSD)范围在0.69%~2.8%之间,表明本试验精密度良好。

四、样品前处理及样品分析
取10g饮料样品(精确称量至0.001g)于50mL具塞离心管中,加适量水,超声15min,用水定容至刻度,经高速离心机(4000r/min)离心10min,再用0.22μm微孔滤膜过滤,滤液供UPLC测定。
在样品上进行5.1μg/mL加标回收率试验,每组进行6次平行测定,并与传统液相色谱法进行对照分析,如表4所示,UPLC检测的各组分样品回收率在97.9%~101.1%之间,所有目标物的相对标准偏差均小于5.0%,且两种方法所得到的结果有良好的一致性。


五、结论
本方法采用超高效液相色谱法测定饮料中10种食品添加剂,在优化后的试验条件下,各组分在10min内可达到完全分离,且具有良好的线性关系,检出限为0.0017~0.0034μg/mL,样品加标回收率为97.9%~101.1%。此方法与传统液相色谱法相比,可大大缩短测定的分析时间,有效提高了检测效率,具有现实指导意义,为应用于食品中添加剂的检测奠定了基础。
声明:本文所用图片、文字来源《中国食品添加剂》,版权归原作者所有。如涉及作品内容、版权等问题,请与本网联系
相关链接:甲醇,乙酸铵,添加剂
- ·剑与远征驭星猎手背景故事与角色技能深度解析
- ·广东深圳:3天查扣质量违法电动自行车623件
- ·新春走基层|秦都人的质量“逐梦”故事
- ·江苏盐城:精准勾画放心消费全景图
- ·【网络中国节·元宵】非遗展演闹元宵
- ·市场监管行风建设在行动
- ·过氧化氢溶液标准物质:高纯度校准,氧化反应实验必备
- ·多糖替代动物脂肪与肌原纤维蛋白相互作用的研究现状(一)
- ·合并,退市!知名车企突传消息
- ·黑龙江大兴安岭地区开展知识产权维权援助专项活动
- ·市场监管文化建设纵览
- ·国家药监局:鹿小新草本祛痘凝胶等39批次化妆品抽检不合格
- ·江湖X汉家江湖战斗机制深度解析:伤害计算与攻防公式全面剖析
- ·山东济南:加强重点工业产品质量监管
- ·吉林长春:用信息化手段提升重点领域安全监管效能
- ·葡萄酒酿造过程中风险因素的分析研究(一)